The goal of NetCoupler is to estimate causal links between a set of -omic (e.g. metabolomics, lipidomics) or other high-dimensional data and an external variable, such as a disease outcome, an exposure, or both. The NetCoupler-algorithm, initially formulated during Clemens’ PhD thesis (Wittenbecher 2017), links a conditional dependency network with an external variable (i.e. an outcome or exposure) to identify network-independent associations between the network variables and the external variable, classified as direct effects.
A typical use case we have in mind would be if a researcher might be interested in exploring potential pathways that exist between a health exposure like red meat consumption, its impact on the metabolic profile, and the subsequent impact on an outcome like type 2 diabetes incidence. So for instance, you want to ask questions to get answers that look like the figure below.
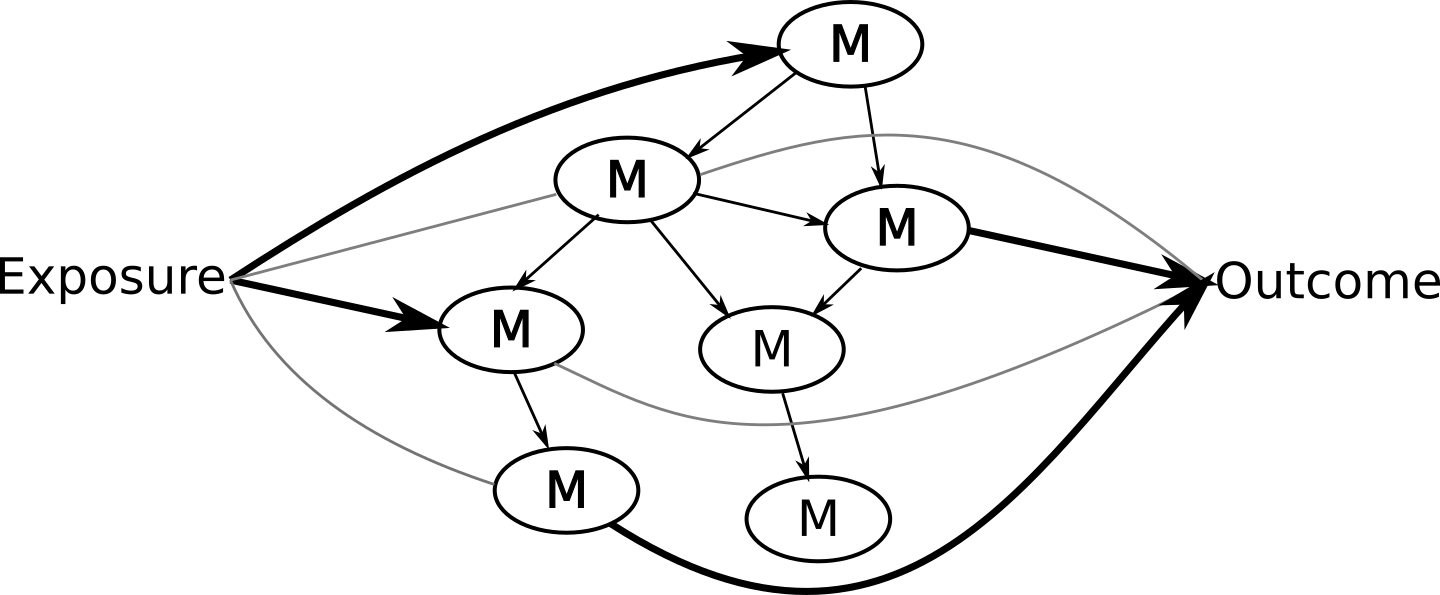
The input for NetCoupler includes:
- Standardized metabolic or other high-dimensional data.
- Exposure or outcome data.
- Network estimating method (default is the PC algorithm from the pcalg package).
- Modeling method (e.g. linear regression with
lm()), including confounders to adjust for.
The final output is the modeling results along with the results from NetCoupler’s classification. Results can then be displayed as a joint network model in graphical format.
There are a few key assumptions to consider before using NetCoupler for your own research purposes.
- -omics data is the basis for the network. We haven’t tested this on non-omics datasets, so can’t guarantee it works as intended.
- The variables used for the metabolic network are numerical
- Metabolic data should have a theoretical network underlying it.
- Missing data are not used in any of the NetCoupler processes.
Overall package framework
NetCoupler has several frameworks in mind:
- Works with magrittr
%>%or base R|>operator. - Works with tidyselect
helpers (e.g.
starts_with(),contains()). - Is auto-complete friendly (e.g. start function names with
nc_). - Inputs and outputs of functions are tibbles/dataframes or tidygraph tibbles.
- Generic modeling approach by using model and settings as function
argument inputs.
- This allows flexibility with what model can be used (e.g. linear regression, cox models).
- Almost all functionality of modeling in R is available here, for instance handling of missing data or of categorical variables.
Workflow
The general workflow for using NetCoupler revolves around several main functions, listed below as well as visualized in the figure below:
-
nc_standardize(): The algorithm in general, but especially the network estimation method, is sensitive to the values and distribution of the variables. Scaling the variables by standardizing, mean-centering, and natural log transforming them are important to obtaining more accurate estimations. -
nc_estimate_network(): Estimate the connections between metabolic variables as a undirected graph based on dependencies between variables. This network is used to identify metabolic variables that are connected to each other as neighbours.- We plan on implementing other network estimators aside from the PC-algorithm at some point in the future.
-
nc_estimate_exposure_links()andnc_estimate_outcome_links(): Uses the standardized data and the estimated network to classify the conditionally independent relationship between each metabolic variable and an external variable (e.g. an outcome or an exposure) as either being a direct, ambiguous, or no effect relationship.- Setting the threshold for classifying effects as direct, ambigious,
or none is done through the argument
classify_option_list. See the help documentation of the estimating functions for more details. For larger datasets, with more sample size and variables included in the network, we strongly recommend lowering the threshold used to reduce the risk of false positives.
- Setting the threshold for classifying effects as direct, ambigious,
or none is done through the argument
-
nc_join_links(): Not implemented yet. Join together the exposure- and outcome-side estimated links. -
nc_plot_network(): Not implemented yet. Visualize the connections estimated fromnc_estimate_network(). -
nc_plot_links(): Not implemented yet. Plots the output results from eithernc_estimate_exposure_links(),nc_estimate_outcome_links(), ornc_join_links().
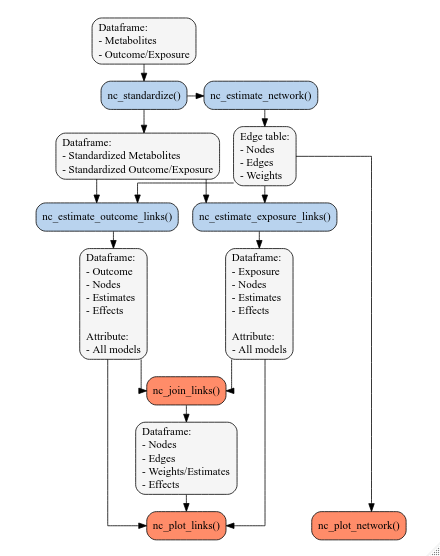
NetCoupler functions and their input and ouput. Input and output objects are the light gray boxes, while the light blue boxes are the currently available functions, and the light orange boxes are functions planned to be developed.
Simple example
The below is an example using a simulated dataset for demonstrating
NetCoupler. For more examples, particularly on how to use with different
models, check out the vignette("examples").
Estimating the metabolic network
For estimating the network, it’s (basically) required to standardize
the metabolic variables before inputting into
nc_estimate_network(). This function also log-transforms
and scales (mean-center and z-score normalize) the values of the
metabolic variables. We do this because the network estimation algorithm
can sometimes be finicky about differences in variable numerical scale
(mean of 1 vs mean of 1000).
library(NetCoupler)
std_metabolic_data <- simulated_data %>%
nc_standardize(starts_with("metabolite"))If you have potential confounders that you need to adjust for during
the estimating links phase of NetCoupler, you’ll need to include these
confounding variables when standardizing the metabolic variables. You do
this by regressing the confounding variables on the metabolic variables
by using the regressed_on argument of
nc_standardize(). This will automatically first standardize
the variables, run models on the metabolic variables that includes the
confounding variables, and then extract the residuals from the model
which are then used to construct the network. Here’s an example:
std_metabolic_data <- simulated_data %>%
nc_standardize(starts_with("metabolite"),
regressed_on = "age")After that, you can estimate the network. The network is by default
estimated using the PC-algorithm. You can read more about it in the help
page of the pc_estimate_undirected_graph() internal
function.
# Make partial independence network from metabolite data
metabolite_network <- std_metabolic_data %>%
nc_estimate_network(starts_with("metabolite"))Estimating exposure and outcome-side connections
For the exposure and outcome side, you should standardize the metabolic variables, but this time, we don’t regress on the confounders since they will be included in the models.
standardized_data <- simulated_data %>%
nc_standardize(starts_with("metabolite"))Now you can estimate the outcome or exposure and identify direct
effects for either the exposure side
(exposure -> metabolite) or the outcome side
(metabolite -> outcome). For the exposure side, the
function identifies whether a link between the exposure and an index
node (one metabolic variable in the network) exists, independent of
potential confounders and from neighbouring nodes (other metabolic
variables linked to the index variable). Depending on how consistent and
strong the link is, the effect is classified as “direct”, “ambiguous”,
or “none”.
In the example below, we specifically generated the simulated data so that the exposure is associated with metabolites 1, 8, and 12. And as we can see, those links have been correctly identified.
outcome_estimates <- standardized_data %>%
nc_estimate_outcome_links(
edge_tbl = as_edge_tbl(metabolite_network),
outcome = "outcome_continuous",
model_function = lm
)
outcome_estimates
#> # A tibble: 12 × 6
#> outcome index_node estimate std_error fdr_p_value effect
#> * <chr> <chr> <dbl> <dbl> <dbl> <chr>
#> 1 outcome_continuous metabolite_1 0.0466 0.0254 0.124 ambiguous
#> 2 outcome_continuous metabolite_10 0.00449 0.0254 0.947 ambiguous
#> 3 outcome_continuous metabolite_11 -0.00700 0.0254 0.912 none
#> 4 outcome_continuous metabolite_12 0.350 0.0242 0 direct
#> 5 outcome_continuous metabolite_2 -0.0280 0.0255 0.424 none
#> 6 outcome_continuous metabolite_3 -0.0936 0.0252 0.000620 direct
#> 7 outcome_continuous metabolite_4 0.0267 0.0256 0.453 ambiguous
#> 8 outcome_continuous metabolite_5 0.103 0.0253 0.000167 ambiguous
#> 9 outcome_continuous metabolite_6 0.113 0.0252 0.0000237 ambiguous
#> 10 outcome_continuous metabolite_7 0.00171 0.0255 0.956 none
#> 11 outcome_continuous metabolite_8 0.0212 0.0253 0.548 none
#> 12 outcome_continuous metabolite_9 0.201 0.0250 0 direct
exposure_estimates <- standardized_data %>%
nc_estimate_exposure_links(
edge_tbl = as_edge_tbl(metabolite_network),
exposure = "exposure",
model_function = lm
)
exposure_estimates
#> # A tibble: 12 × 6
#> exposure index_node estimate std_error fdr_p_value effect
#> * <chr> <chr> <dbl> <dbl> <dbl> <chr>
#> 1 exposure metabolite_1 0.173 0.0228 0 direct
#> 2 exposure metabolite_10 0.318 0.0219 0 direct
#> 3 exposure metabolite_11 0.0543 0.0232 0.0409 ambiguous
#> 4 exposure metabolite_12 0.0242 0.0231 0.380 none
#> 5 exposure metabolite_2 -0.0430 0.0231 0.106 ambiguous
#> 6 exposure metabolite_3 0.0411 0.0231 0.123 ambiguous
#> 7 exposure metabolite_4 0.00344 0.0232 0.920 none
#> 8 exposure metabolite_5 0.0479 0.0232 0.0717 ambiguous
#> 9 exposure metabolite_6 -0.0189 0.0230 0.506 none
#> 10 exposure metabolite_7 -0.162 0.0229 0 direct
#> 11 exposure metabolite_8 -0.355 0.0216 0 direct
#> 12 exposure metabolite_9 0.0571 0.0230 0.0292 ambiguousIf you want to adjust for confounders and have already used
regressed_on in the nc_standardize() function,
add confounders to nc_estimate_outcome_links() or
nc_estimate_exposure_links() with the
adjustment_vars argument:
outcome_estimates <- standardized_data %>%
nc_estimate_outcome_links(
edge_tbl = as_edge_tbl(metabolite_network),
outcome = "outcome_continuous",
model_function = lm,
adjustment_vars = "age"
)Slow code? Use parallel processing with future
If the analysis is taking a while, you can use the future package to
speed things up by implementing parallel processing. It’s easy to use
parallel processing with NetCoupler since it uses the future package. By
setting the “processing plan” with future::plan() to
multisession, NetCoupler will use parallel processing for
its computationally intensive component of the algorithm. After you run
your code, close up the parallel processing by putting it back to normal
with plan(sequential). Using the future package you can
speed up the processing by almost 2.5 times.
# You'll need to have furrr installed for this to work.
library(future)
plan(multisession)
outcome_estimates <- standardized_data %>%
nc_estimate_outcome_links(
edge_tbl = as_edge_tbl(metabolite_network),
outcome = "outcome_continuous",
model_function = lm
)
plan(sequential)